What does a genetic variant actually mean for the person who carries it?
We move past binary “pathogenic / benign” labels to give each carrier a calibrated probability of a clinically meaningful outcome — pairing high-throughput biology, protein structure, and Bayesian modeling.
A variant is rarely simply benign or pathogenic. We estimate the probability it leads to a concerning clinical outcome.
Each variant gets a calibrated, individualized estimate — far more actionable for a clinician or a family than a single categorical label.
From a binary label to a calibrated probability.
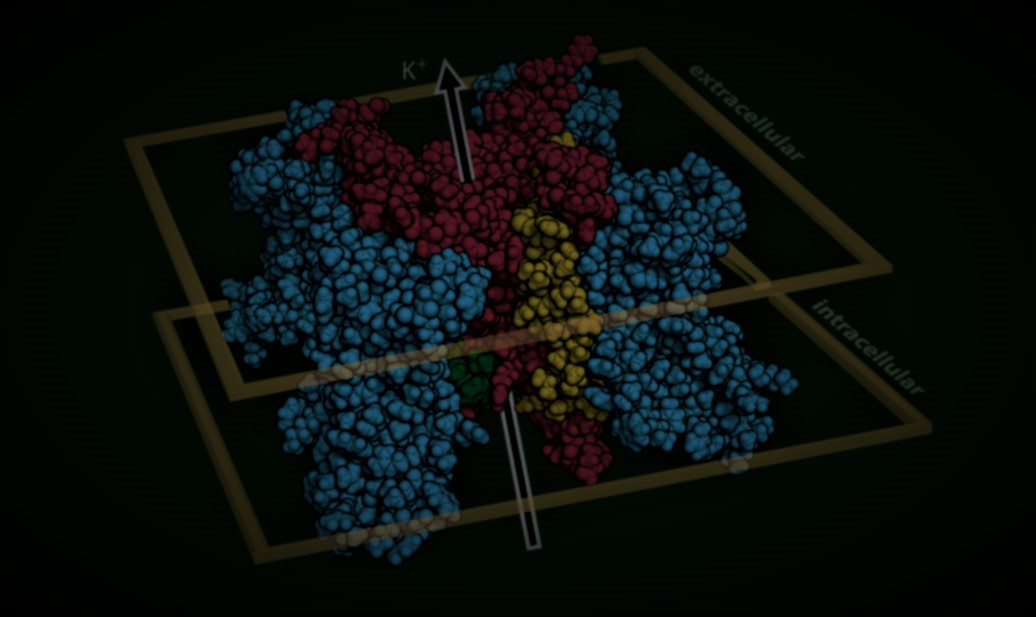
An open-source pipeline mines the literature with large language models, unifies AlphaMissense, REVEL, CADD, ClinVar and gnomAD, folds in 3D structural priors, and returns penetrance with explicit uncertainty — for cardiac arrhythmia genes like KCNH2 and SCN5A.
Read our approach →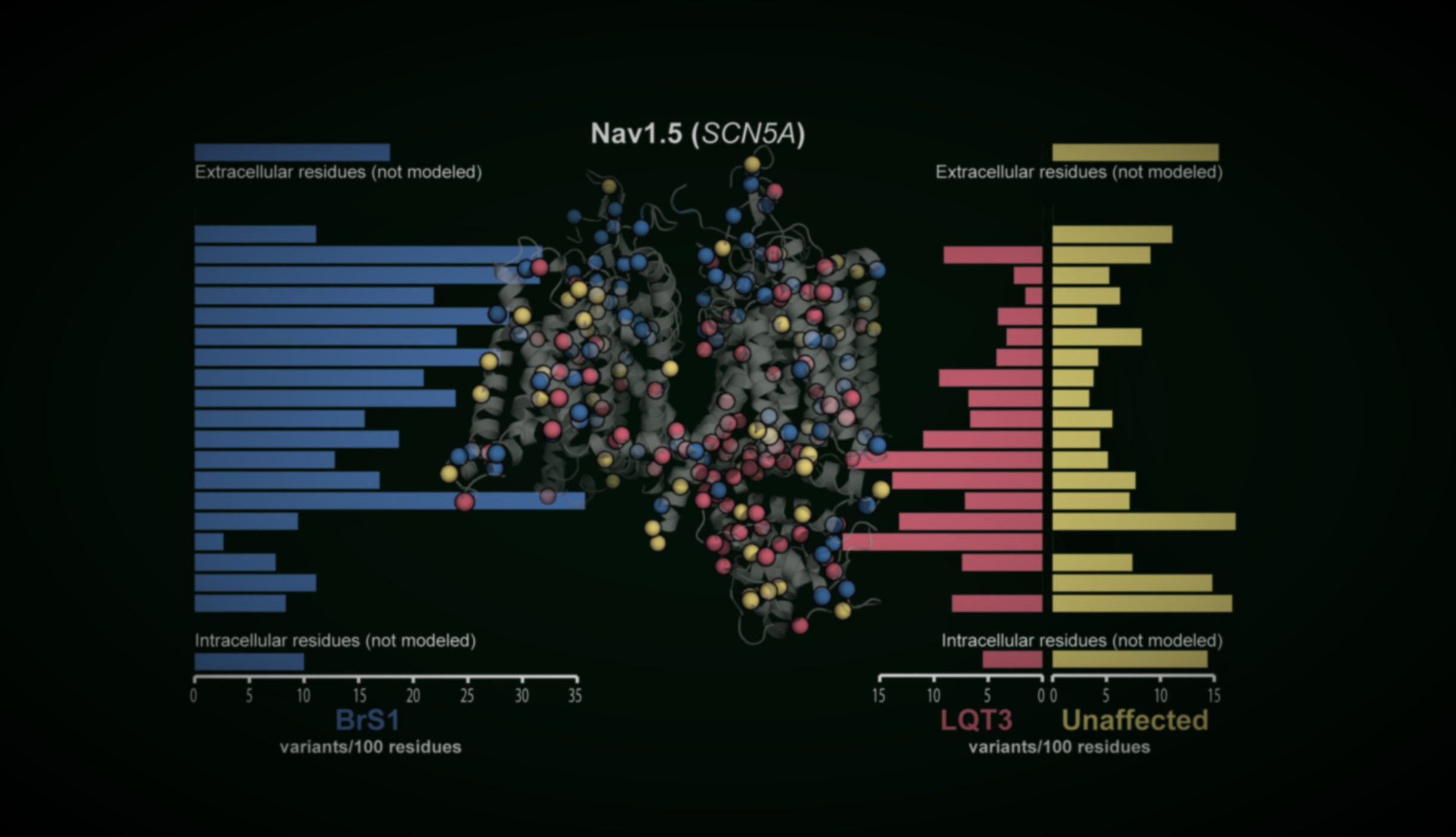
Four steps from the published record to a calibrated risk estimate.
Curate
LLM agents pull per-variant carrier counts, phenotypes and study context from PubMed; a multi-model consensus scores gene–disease validity against the ClinGen SOP.
Annotate
AlphaMissense, REVEL, CADD, ClinVar and gnomAD are unified into one queryable resource, so every variant carries the same panel of evidence.
Localize
Variants are mapped onto AlphaFold structures with elastic-network analysis, so pathogenic clusters raise the prior for nearby variants of uncertain significance.
Estimate
A Bayesian model fuses carriers, predictive features and structural priors into penetrance estimates with full uncertainty — our patented framework.
What we are working on now.
LLM-assisted curation
Pipelines mine PubMed for variant, phenotype and carrier evidence, then a multi-model consensus scores gene–disease validity.
Multi-source features
AlphaMissense, REVEL, CADD, ClinVar and gnomAD unified into one resource, so every variant carries the same evidence.
Structure-aware priors
AlphaFold models place variants in 3D, letting pathogenic neighborhoods inform priors for nearby VUS.
Bayesian penetrance
A probabilistic model fuses carrier counts, features and structural priors into penetrance with full uncertainty.
High-throughput function
Deep mutational scanning and automated patch clamp measure variant effects at scale — including the fullest KCNH2 map to date.
Stem-cell & clinical
CRISPR-edited iPSC-cardiomyocytes test how polygenic background shifts expressivity; survival models flag breakthrough events.
VariantBrowser.org
A freely available portal used by clinicians, genetic counselors and researchers worldwide. Each gene page unifies carrier counts, functional data, structural context and in silico predictors behind a calibrated Bayesian estimate of disease probability.
Explore the browser →