How do we determine the significance of genetic variants?
Integrating in vitro and in silico evidence to quantify disease probability — rather than relying on binary pathogenicity labels.
What is the significance of a genetic variant to those who carry it?
Next-generation sequencing has revealed countless variants across the genome, yet translating those observations into patient-level insight remains hard, especially for rare alleles. We resolve variants of uncertain significance by integrating in vitro and in silico evidence to quantify disease probability rather than relying on binary labels.
We begin with cardiac ion-channel genes implicated in long QT syndrome, Brugada syndrome and related arrhythmias because they offer a tractable system for connecting molecular mechanism to clinical outcome. For an overview, watch this talk.
curated
unified
per curation
VariantBrowser
An integrated, open-source path from literature to penetrance.
Curate
LLM agents pull carrier counts, phenotypes and study context from PubMed; multi-model consensus scores gene–disease validity against the ClinGen SOP.
Annotate
AlphaMissense, REVEL, CADD, ClinVar and gnomAD unified into one database, so every variant carries the same panel of predictive evidence.
Localize
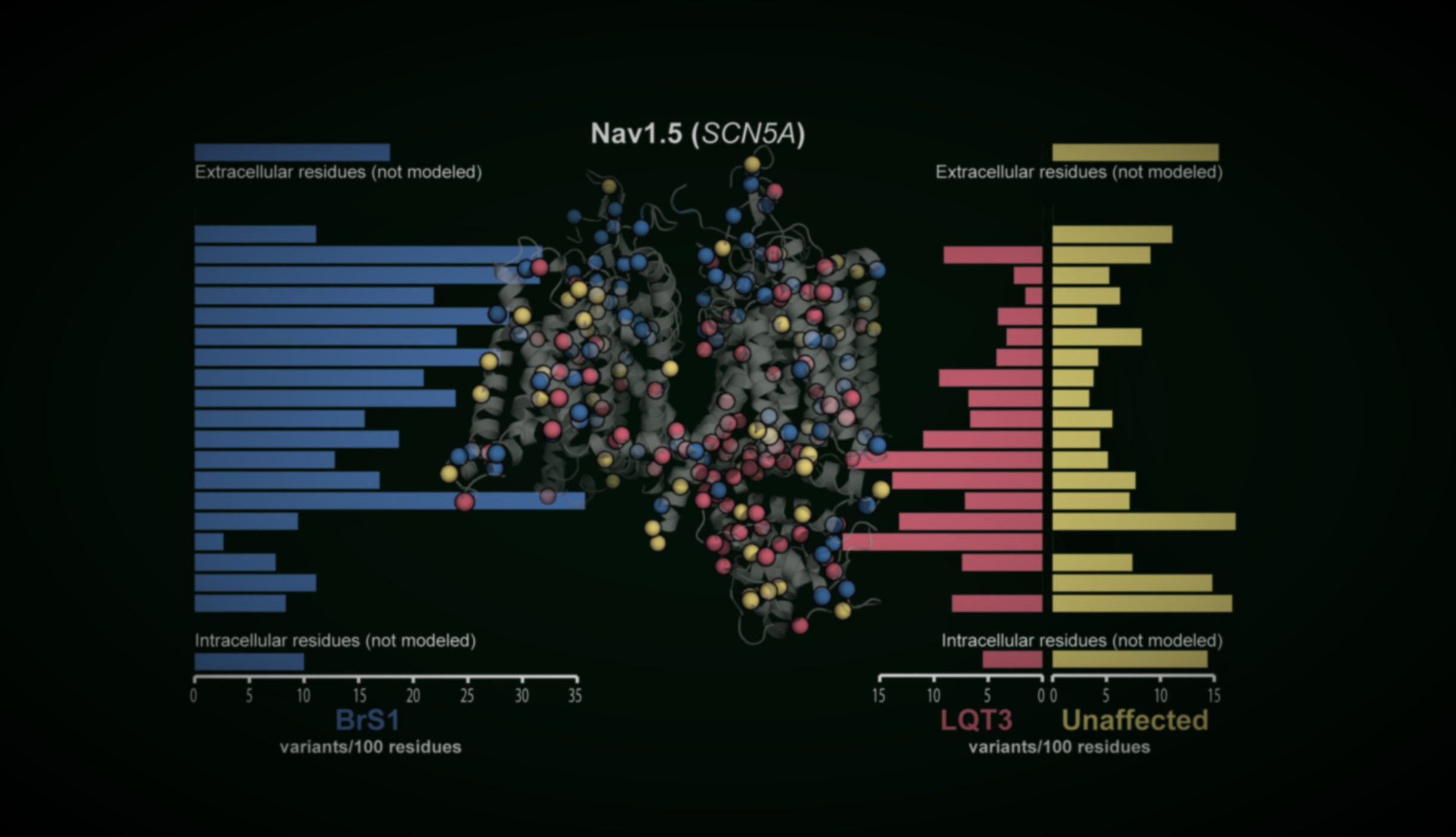
Variants mapped onto AlphaFold structures with elastic-network analysis, so pathogenic clusters raise the prior for nearby VUS.
Estimate
A Bayesian model fuses carriers, features and structural priors into penetrance with full uncertainty — our patented method.
Three advances we've built the program on.
Functional genomics + modeling
We pioneered deep mutational scanning and scalable automated patch clamp to interpret ion-channel variants — data that feeds models estimating penetrance for individual carriers.
Bayesian penetrance estimation
Our patented framework incorporates functional data, clinical observations and genetic background to refine individualized risk estimates.
Literature & gene–disease curation
Large language models extract variant, phenotype and carrier evidence at scale; a multi-model consensus reproduces ClinGen-style validity calls in an auditable workflow.
VariantBrowser.org
Each gene page unifies published carrier counts, functional data, structural context and in silico predictors behind a calibrated Bayesian estimate of disease probability.
Four cardiac channelopathy genes are live today — together spanning more than 55,000 variants — with many more in active development.
Explore VariantBrowser.org →How much functional perturbation is too much?
By curating more than 1,400 SCN5A variants — including 304 with functional data — we show that modest perturbations yield heterogeneous clinical presentations, whereas extreme loss or gain of function produces more consistent outcomes. Modeling disease probability from functional effect offers a clearer path to clinical decisions than binary classification.
Intravariant heterogeneity
Carriers of the same variant can present very differently. Peak current — a proxy for channel function — tracks with, but does not fully determine, how many carriers are diagnosed with Brugada syndrome. Capturing that spread is exactly what our penetrance models are built to do.
| Variant | Peak current | # unaffected | # BrS1 |
|---|---|---|---|
| S1787N | 95% | 12 | 1 |
| Y1795H | 66% | 7 | 5 |
| R367H | 0% | 3 | 16 |
Peak current = proxy for channel function · unaffected = carriers without phenotype · BrS1 = carriers diagnosed with Brugada syndrome.
Blending experiment and computation.
We interrogate structural consequences with Rosetta modeling, molecular dynamics in AMBER and NMR, and generate functional measurements through deep mutational scanning and automated patch clamp. These complementary approaches let us build predictive models we can validate in disease-relevant systems.
Measuring variant effects at scale.
Interpreting variants one at a time cannot keep pace with sequencing. We characterize thousands of variants functionally and feed them directly into our penetrance models.
Deep mutational scanning
The most comprehensive variant-effect map to date for KCNH2/Kv11.1 — cell-surface trafficking for thousands of variants in one experiment.
Automated patch clamp
Scalable electrophysiology measures peak current, gating and kinetics far faster than manual recording.
iPSC-cardiomyocytes
CRISPR-edited stem-cell cardiomyocytes with defined backgrounds test how polygenic variation reshapes rare-variant expressivity.
Polygenic risk scores modify susceptibility. In recent work (2026) we used quantitative proteomics to show how elevated QT polygenic risk reshapes Kv11.1 (hERG) trafficking networks in cardiomyocytes — connecting common-variant background to a concrete molecular mechanism, and revealing nonlinear interactions that influence arrhythmia risk and treatment response.
Atrial fibrillation
The framework is gene-agnostic. A current pilot brings our LLM-assisted, ClinGen-style gene–disease validity curation to atrial fibrillation — starting with NPPA, GJA5, KCNA5 and PITX2 — in a transparent, reviewable workflow that can scale to many more genes.
Breakthrough events
Knowing a variant is pathogenic is not the same as knowing whether a patient will do well on treatment. Using harmonized international cohorts, we build survival models of breakthrough cardiac events — those occurring despite beta-blocker therapy — in KCNH2 carriers, to flag individuals who need more aggressive management before an event occurs.
Our research is supported by NIH R01HL160863 (2022–2027), NIH K99/R00 HL135442 (2017–2022), and the American Heart Association Career Development Award (2021–2024).


